
|
Through-Space Interaction Mediated by a Sulfoxide By Joel Meyer, G.; Smith, Elliott R.; Sakamoto, Takahiro; Lichtenberger, Dennis L.; Glass, Richard S.
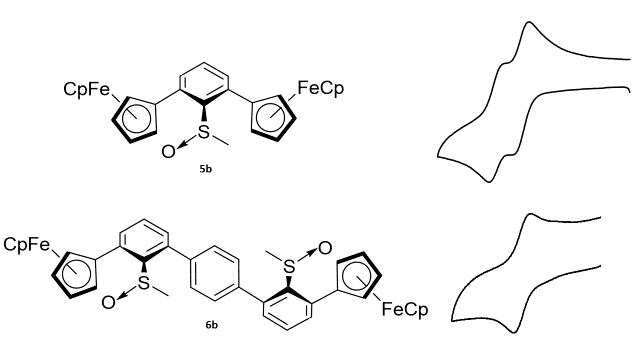
Two new bisferrocenylphenylsulfoxides were synthesized and studied to det. the effect of the polar sulfoxide bond on through-space interaction between ferrocene moieties. The electronic and redox properties of these compds. were studied by UPS, cyclic voltammetry, differential pulse voltammetry, and d. functional theory computations. Electrochem. results for 2,6-bis(ferrocenyl) thioanisole S-oxide(I) show two, fully reversible one-electron redox processes. The initial oxidn. shows a 62-mV neg. shift compared with the sulfide analog 2,6-bis(ferrocenyl)thioanisole, and an increased peak sepn. for the oxidn. of 160 vs. 145 mV. No peak sepn. is obsd. in II. No intervalence charge transfer band was obsd. in the complex I1+ by UV-Visible/Near-IR spectroscopy, ruling out electronic communication. Thus, the through-space electrostatic interactions of the sulfoxide renders the non-equiv. ferrocenes in I to have different oxidn. potentials.
|
|
From gas-phase ionization energies to solution oxidation potentials: Dimolybdenum tetraformamidinate paddlewheel complexes By Van Dorn, Laura O.; Borowski, Susan C.; Lichtenberger, Dennis L.
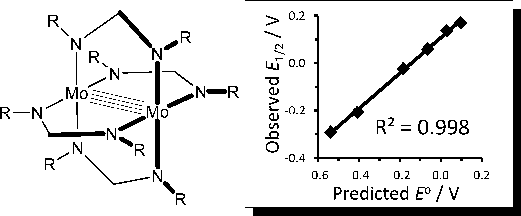
The gas-phase ionization energies of a series of Mo2(DPhF)4 paddlewheel complexes (DPhF is the N,N'-diphenylformamidinate anion with p-CH3, p-Cl, m-Cl, p-CF3, or m-CF3 Ph substituents) have been measured by UPS and compared with the soln. oxidn. potentials measured by cyclic voltammetry (CV) reported by Ren and coworkers. A linear relationship was found between the gas-phase ionization energies and the soln. oxidn. potentials. D. functional theory (DFT) computations clarify the individual electronic and thermodn. factors that contribute to the correlation. The metal-metal delta bond electron energy is the largest factor in detg. the soln. oxidn. potential. The substituents shift the metal-metal orbital energies by changing the through-space field potential at the metals rather than by an inductive change in charge at the metals or orbital overlap effects. The cation solvation energies det. the extent that the potential shifts are attenuated in soln. The results show that substituent field effects and solvation have major roles in detg. the dimetal redox chem. even when the dimetal unit is protected from direct interaction with the substituent and the solvent.
|
|
Phosphine-substituted (η5-pentadienyl) manganese carbonyl complexes: Geometric structures, electronic structures, and energetic properties of the associative substitution mechanism, including isolation of the slipped η3-pentadienyl associative intermediate de la Cruz Cruz, Jose Ignacio, Patricia Juarez-Saavedra, Brenda Paz-Michel, Marco Leyva-Ramirez, Asha Rajapakshe, Aaron K. Vannucci, Dennis L. Lichtenberger and M. Paz-Sandoval. Organometallics, 2014, 33, 278-288. (http://dx.doi.org/10.1021/om401017t)
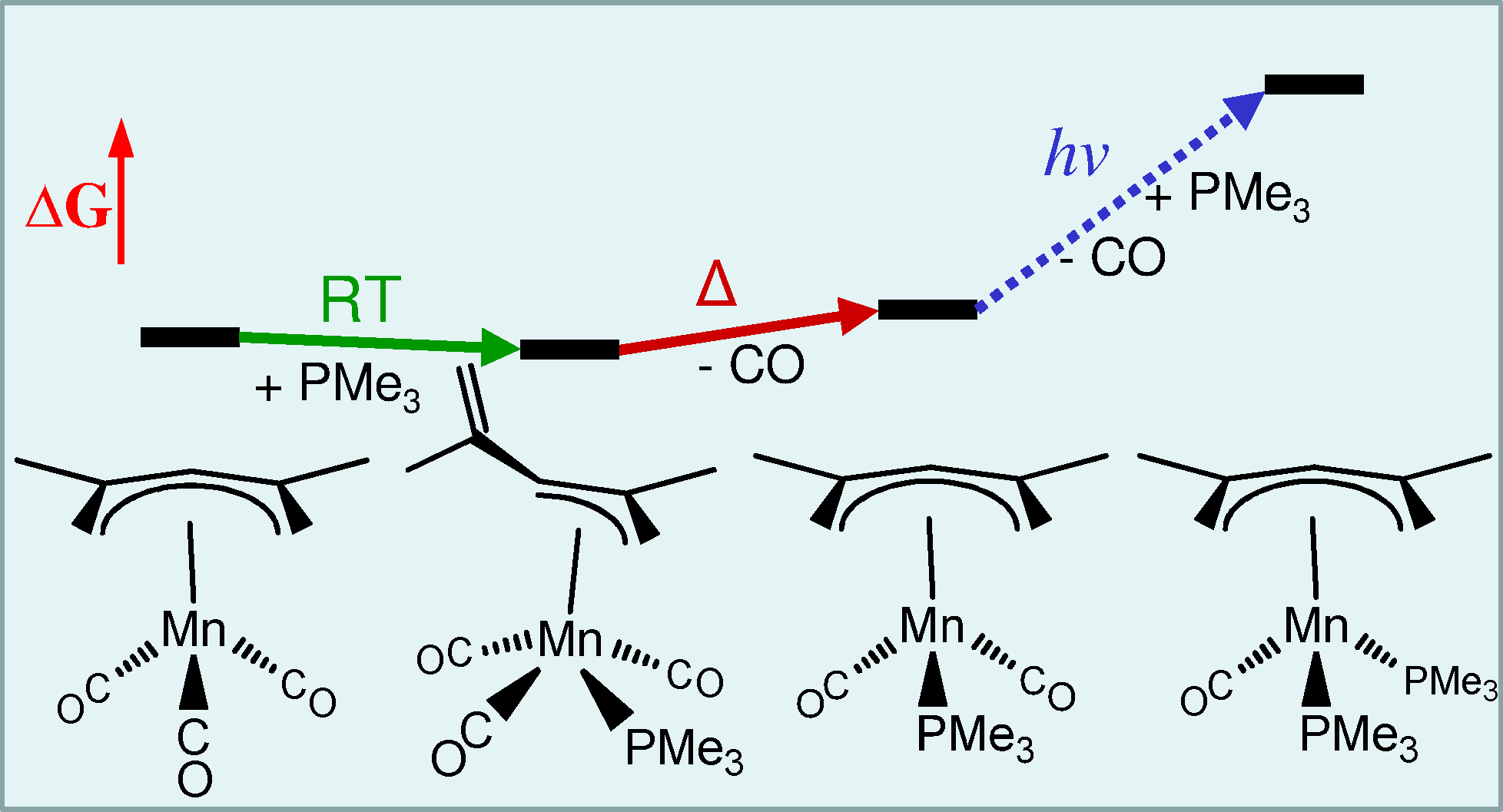
The molecule (h5-Me2Pdl)Mn(CO)3 (h5-Me2Pdl = 2,4-dimethyl-η5-pentadienyl) has been prepared by a new method and used as a starting material to prepare the molecules (h5‑Me2Pdl)Mn(CO)n(PMe3)3-n (n = 2, 1) by phosphine substitution for carbonyls. The first carbonyl substitution is achieved thermally in refluxing cyclohexane, and the second carbonyl substitution requires photolysis. At room temperature in benzene the associative intermediate (h3-Me2Pdl)Mn(CO)3(PMe3) that precedes the initial loss of carbonyl is observed. Single crystal structures are reported for all complexes, including the associative intermediate of the first substitution in which the pentadienyl ligand has slipped to the η3 bonding mode. These molecules offer an opportunity to examine fundamental principles of the interactions between metals and pentadienyl ligands compared to the well-developed chemistry of metal-cyclopentadienyl (Cp) complexes as a function of electron richness at the metal center. Photoelectron spectra of these molecules show that the Me2Pdl ligand has π ionizations at lower energy than the analogous Cp ligand and donates more strongly to the metal than the Cp ligand, making the metal more electron rich. Phosphine substitutions for carbonyls further increase the electron richness at the metal center. Density functional calculations provide further insight into the electronic structures and bonding of the molecules, revealing the energetics and role of the pentadienyl slip from η5 to η3 bonding in the early stage of the associative substitution mechanism. Computational comparison with dissociative ligand substitution mechanisms reveals the roles of dispersion interaction energies and the entropic free energies in the ligand substitution reactions.
|
|
Solubilizing the Most Easily Ionized Molecules and Generating Powerful Reducing Agents Gina M. Chiarella, F. A. Cotton, Jason C. Durivage, Dennis L. Lichtenberger and Carlos A. Murillo. J. Am. Chem. Soc., 2013, 135, 17889-17896. (http://dx.doi.org/10.1021/ja408291k)
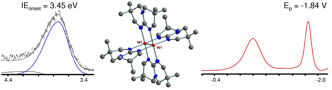
Electron transfer reactions represent one of the most important and ubiquitous processes in Chemistry and they are also essential to sustain life. Stable, strong oxidizing and reducing agents have many important chemical and material applications. Especially needed are strong redox agents to be utilized in non-aqueous, homogeneous systems and for reactions in which stoichiometric control is critical. Two very soluble compounds having W2(bicyclic guanidinate)4 paddlewheel structures show record low ionization energies and very negative oxidation potentials in THF (–1.84 to –1.90 V vs Ag/AgCl). These compounds are thermally stable and easy to synthesize in high yields and good purity. They are very reactive and potentially useful reducing agents in non-polar, non-protonated solvents.
|
|
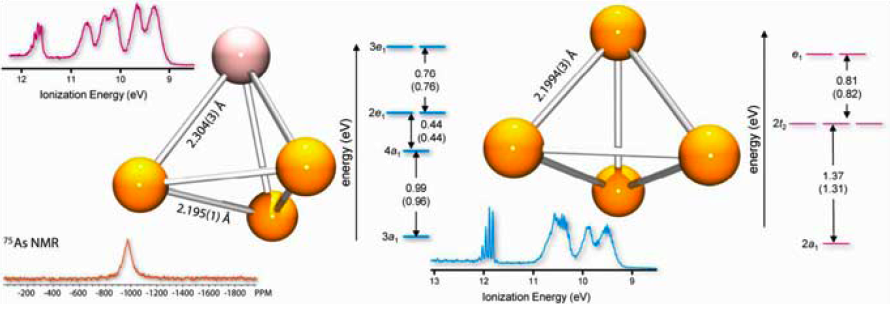
On the Molecular and Electronic Structures of AsP3 and P4 Brandi M. Cossairt, Christopher C. Cummins, Ashley R. Head, Dennis L. Lichtenberger, Raphael J. F. Berger, Stuart A. Hayes, Norbert W. Mitzel, Gang Wu. J. Am. Chem. Soc., 2010, 132 (24), pp 8459–8465. DOI: 10.1021/ja102580d
Recently the group at MIT discovered a directed metal-organic synthetic pathway for the synthesis of the binary inter-pnictogen compound AsP3. AsP3 was found to be thermally stable over a wide temperature range and the compound can be isolated in pure form. Due to its structural simplicity, thermal stability, heavy-atom composition, and volatility, AsP3 is an ideal candidate for investigation by gas-phase electron diffraction (GED) and by gas-phase photoelectron spectroscopy, unbiased by solid state intermolecular interactions. The electronic structures of neutral AsP3 and P4, and the energies associated with geometric distortion upon ionization of each substance were probed by photoelectron spectroscopy. Solid-state nuclear magnetic resonance (NMR) investigations were provided to complement the gas-phase measurements by direct interrogation of the electronic environments of the arsenic atom (75As NMR) and the phosphorus atoms (31P NMR) in the solid state. Finally, quantum chemical methods were used to provide a more complete understanding of the electronic structure of AsP3 through spherical aromaticity assessment via gauge-including magnetically induced current (GIMIC) calculations.
|
|
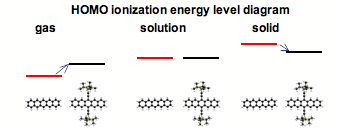
Electronic Properties of Pentacene versus Triisopropylsilylethynyl-Substituted Pentacene: Environment-Dependent Effects of the Silyl Substituent. Olga Lobanova Griffith, John E. Anthony, Adolphus G. Jones, and Dennis L. Lichtenberger, J. Am. Chem. Soc., 2010, 132(2), 580-586.(http://dx.doi.org/10.1021/ja906917r).
Electronic devices based on pentacenes represent a leading next-generation organic electronic device technology. One of the most popular pentacene derivatives is triisopropylsilylethynyl-substituted (TIPS) pentacene which has properties appropriate for its use in thin-film transistors. Energy measures of the intra- and intermolecular electronic effects of the TIPS substituent were obtained from the combination of closely related gas phase and solid phase ultraviolet photoelectron spectroscopy (UPS) measurements along with solution electrochemical measurements. The results showed that the trend in ionization energies with this substitution is reversed going from the single molecule case (gas phase) to the many molecules case (solution, solid phases), with the concomitant changes in charge injection barriers, HOMO-LUMO energy gaps, and charge transport behaviors. The principles that emerged from this analysis were supported by electronic structure calculations at the density functional theory level. The relation between the gas phase and solid phase UPS measurements illustrated in this work provided a general approach to investigating the electronic effects acting on molecules in the solid phase, which in this case are greater than the direct substituent electronic effects within the molecule.
|
|
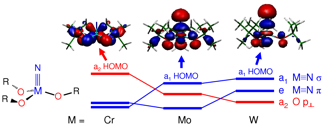
Theoretical and Spectroscopic Investigations of the Bonding and Reactivity of (RO)3M≡N Molecules,
Shentan Chen, Malcolm H. Chisholm, Ernest R. Davidson, Jason B. English and Dennis L. Lichtenberger Inorg. Chem.2009, 48 (3), pp 828–837 (chosen for the cover article). (http://dx.doi.org/10.1021/ic801786u)
The electronic structures of the molecules (tBuO)3M≡N (M = Cr, Mo, W) have been investigated with gas phase photoelectron spectroscopy and density functional calculations. It was found that the alkoxide orbitals mix strongly with the M≡N triple bond orbitals and contribute substantially to the valence electronic structure. The first ionization of (tBuO)3Cr≡N is from an orbital of a2(C3v) symmetry that is oxygen based and contains no metal or nitrogen character by symmetry. In contrast, the first ionizations of the molybdenum and tungsten analogs are from orbitals of a1 and e symmetry that derive from the highest occupied M≡N σ and π orbitals mixed with the appropriate symmetry combinations of the oxygen p orbitals. In this a1 orbital, the oxygen p orbitals mix with the highest occupied M≡N orbital of σ symmetry. This mixing reduces the metal character, consequently reducing the metal-nitrogen overlap interaction in this orbital. From computational modeling, the polarity of the M≡N bond increases down the group such that W≡N has the highest charge separation. In addition to investigation of the effects of the metals, the electronic influences of substitution at the alkoxide ligands have been examined for the molecules (RO)3Mo≡N (R=C(CH3)2H, C(CH3)3, and C(CH3)2CF3). The introduction of CF3 groups stabilizes the molecular orbital energies and increases the measured ionization energies, but does not alter the overall electronic structure. The bonding characteristics of the (tBuO)3M≡N series were compared with those of organic nitriles.
|
|
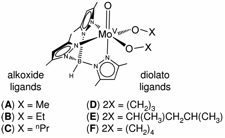
New Insights into Solvolysis and Reorganization Energy from Gas-Phase, Electrochemical, and Theoretical Studies of Oxo-Tp*Mo(V) Compounds. Aaron K. Vannucci, Rae Ana Snyder, Nadine E. Gruhn, Dennis L. Lichtenberger, and John H. Enemark, Inorg. Chem.2009, 48(18), 8856-8862.(http://dx.doi.org/10.1021/ic9011058)
Complexes of the general form Tp*MoO(OX)2 (where Tp* = hydrotris(3,5-dimethyl-1-pyrazolyl)borate and (OX)2 = (OMe)2, (OEt)2, and (OnPr)2 for alkoxide ligands, and (OX)2 = O(CH2)3O, O(CH2)4O, and O[CH(CH3)CH2CH(CH3)]O for diolato ligands) were studied using gas-phase photoelectron spectroscopy, cyclic voltammetry, and density functional theory calculations to examine the effect of increasing ligand size on the oxo-molybdenum core. Oxidation potentials and first ionization energies were shown to be highly sensitive to the number of carbon atoms present in the diolato and alkoxide ligands. A linear correlation between the solution-phase oxidation potentials and the gas-phase ionization energies resulted in an unexpected slope of greater than unity. Density functional theory calculations indicated that cation reorganization energies ranged from 0.15 – 0.51 eV, and this unique correlation was a result of the large differences in the cation reorganization energies.
|
|
Electron Transfer Parameters of Triisopropylsilylethynyl-Substituted Oligoacenes Olga Lobanova Griffith, Nadine E. Gruhn, John E. Anthony, Balaji Purushothaman and Dennis L. Lichtenberger J. Phys. Chem. C, 2008, 112 (51), 20518–20524. (http://dx.doi.org/10.1021/jp8070629)
Understanding the electronic properties and electron transfer characteristics of functionalized oligoacenes is of great importance for the fabrication of organic electronic devices. Charge transfer parameters of bis-triisopropylsilylethynyl-substituted anthracene, tetracene, and pentacene were investigated based on the analysis of their ionization energies and radical cation reorganization energies. High-resolution gas-phase photoelectron spectroscopy measurements and first-principles quantum-mechanical calculations at the density functional theory level were performed. The results indicated that the ionization energies in the gas phase and the inner-sphere reorganization energies are sensitive to the number of fused rings and the substitution by the triisopropylsilylethynyl (TIPS) group. This TIPS functional group shifts the first ionization band of these molecules to lower energy in the gas phase due to mixing between the frontier occupied orbitals of the TIPS group with the highest occupied acene orbital. This mixing adds geometry modifications of the TIPS substituents to those of the acene core that occur with ionization, resulting in a near doubling of the reorganization energies with electron transfer for these molecules.
|

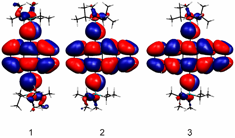
Current and Past Collaborators, Photoelectron Spectroscopy
| John E. Anthony | University of Kentucky |
| Neal R. Armstrong | The University of Arizona |
| Michael T. Ashby | University of Oklahoma |
| Andrew R. Barron | Rice University |
| Fred Basolo (deceased) | Northwestern University |
| Eric Block | State University of New York, Albany |
| Jesse L. Beauchamp | California Institute of Technology |
| Jean-Luc Bredas | King Abdullah University of Science and Technology, Saudi Arabia |
| R. Morris Bullock | Pacific Northwest National Laboratories |
| Bruce E. Bursten | Worcester Polytechnic Institute |
| Nicolai Burzlaff | University Erlangen-Nurnberg, Germany |
| Kenneth G. Caulton | Indiana University |
| Malcolm H. Chisholm | The Ohio State University |
| Brandi M. Cossairt | University of Washington |
| F. Albert Cotton (deceased) | Texas A&M University |
| Christopher C. Cummins | Massachusetts Institute of Technology |
| Lawrence F. Dahl | University of Wisconsin |
| Donald J. Darensbourg | Texas A&M University |
| Evgeny V. Dikarev | University of Albany |
| John H. Enemark | The University of Arizona |
| Richard D. Ernst | University of Utah |
| Dennis H. Evans | Purdue University |
| William J. Evans | University of California, Irvine |
| William E. Geiger | University of Vermont |
| John A. Gladysz | Texas A&M University |
| Richard S. Glass | The University of Arizona |
| Harry B. Gray | California Institute of Technology |
| Robert H. Grubbs | California Institute of Technology |
| Joseph A. Heppert | The University of Kansas |
| Wolfgang A. Herrmann | Technical University Munchen |
| Soren V. Hoffmann | Aarhus University, Denmark |
| Michael D. Hopkins | University of Chicago |
| Donald R. Huffman | The University of Arizona (Physics) |
| Russel P. Hughes | Dartmouth University |
| Nikola C. Jones | Aarhus University, Denmark |
| Antoine Kahn | Princeton University |
| Martin L. Kirk | University of New Mexico |
| Louis Y. Kuo | Lewis and Clark College |
| Mikhail A. Kuznetsov | Saint Petersburg State University, Russia |
| Carlos A. Murillo | Texas A&M University |
| Michael H. Palmer | University of Edinburgh, Scotland, United Kingdom |
| Maria de los Angeles Paz-Sandoval | Centro de Investigacion y de Estudio Avanzados del Instituto Politécnico Nacional, Mexico |
| Jeffrey L. Petersen | University of West Virginia |
| David E. Richardson | University of Florida |
| Robert R. Rye | Sandia National Laboratories |
| Dror Sarid | The University of Arizona (Optical Sciences) |
| Alfred P. Sattelberger | Argonne National Laboratory |
| Wolfdieter A. Schenk | University of Wurzburg, Germany |
| John P. Selegue | University of Kentucky |
| Dieter Sellmann (deceased) | University of Erlangen, Germany |
| Christopher D. Tagge | Occidental College |
| James W. Taylor | University of Wisconsin |
| Ignacio Vargas-Baca | McMaster University |
| Peter C. Vollhardt | University of California Berkeley |
| F. Ann Walker | The University of Arizona |
| Richard A. Walton | Purdue University |
| John H. Weaver | University of Minnesota (Chem. Eng.) |
| Barry L. Westcott | Central Connecticut State University |
| Wolfgang Weigand | University of Jena, Germany |
| Thomas H. Whitesides | University of Wisconsin |
| Jay R. Winkler | California Institute of Technology |
| Fred Wudl | University of California Santa Barbara |
| Vicki H. Wysocki | The Ohio State University |